Quantum Chemical Modelling
Predict and optimise reactions for fluids, minerals and at mineral–fluid interfaces.
We apply quantum mechanical methods (DFT) to quantify speciation, adsorption, and reaction energetics, turning atomic‑scale insights into actionable process recommendations.
Key Questions We Answer
- Which ligands stabilise target metals under given temperature, pressure, and pH?
- How strongly do complexes bind to specific mineral surfaces or defects?
- Do impurities or redox changes alter reactivity or phase stability?
- What structural or vibrational changes occur during substitution?
diopside-water interface with explicit water molecules
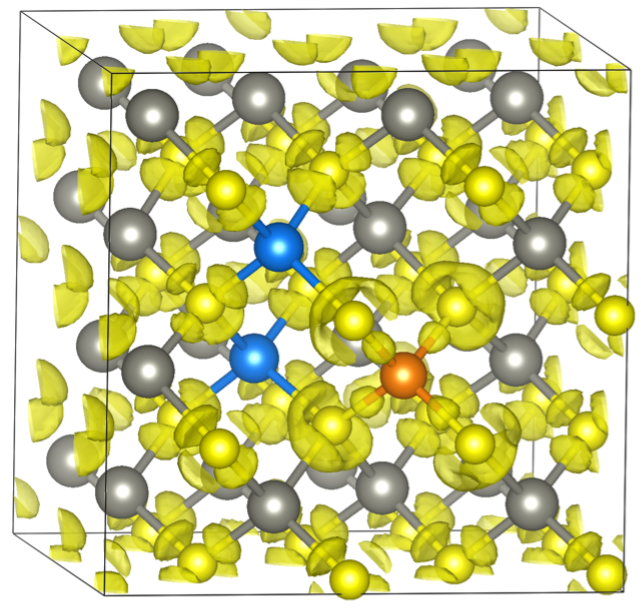
Snapshots of optimized geometry for Ge and Cu incorporation in sphalerite. (Ge=orange, Cu=blue, Zn=grey, S=yellow, yellow cloud shows electron density)
What We Deliver
- Binding and adsorption energies with uncertainty ranges
- Optimised structures and electron density maps
- Surface complexation constants and speciation inputs for thermodynamic models
- Defect/redox sensitivity analyses for process optimisation
- Figures and summaries ready for reports and proposals
Our Approach
- DFT and AIMD simulations using slab and cluster models
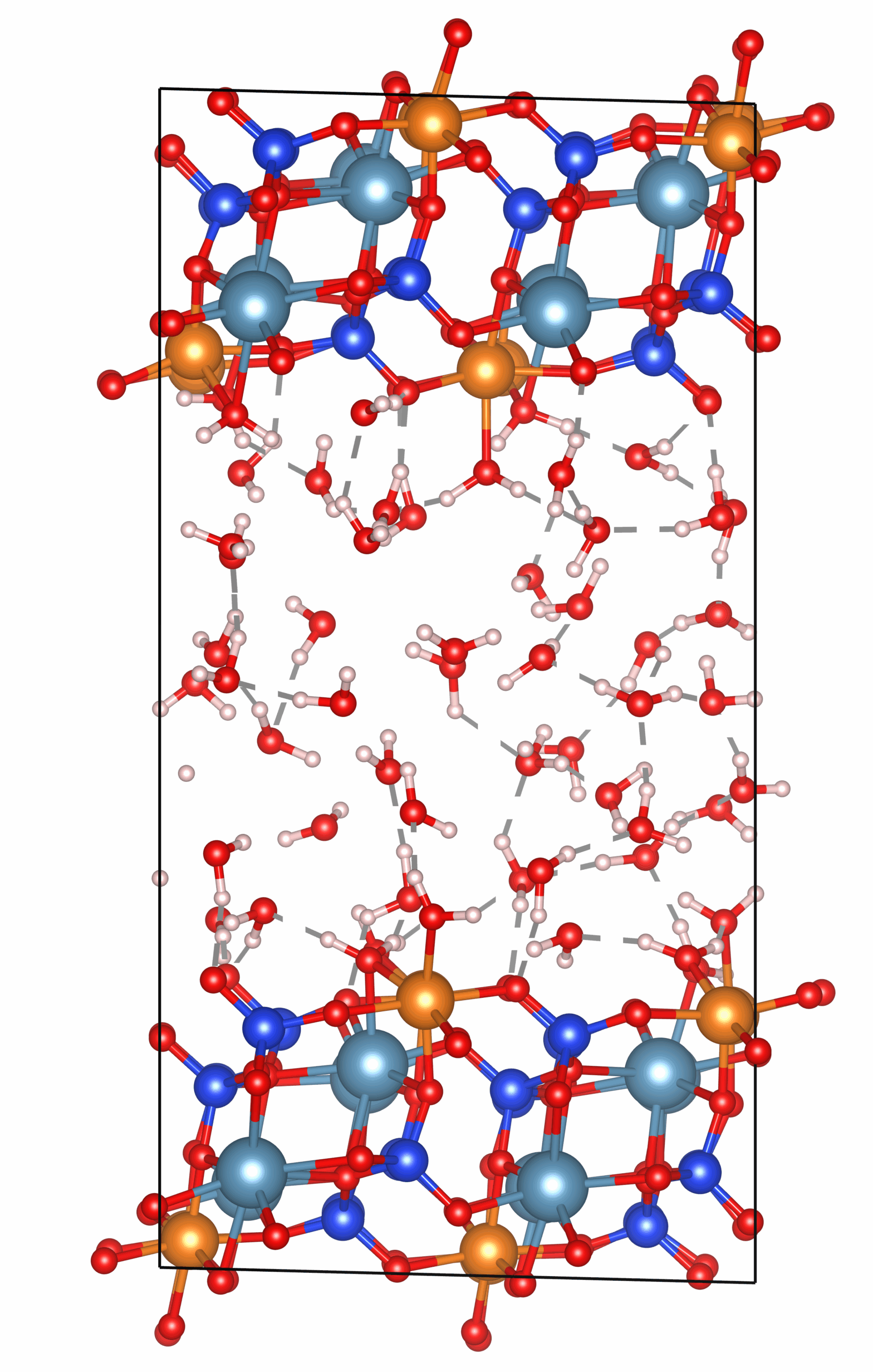
- Explicit/implicit solvation for realistic fluid conditions
- Defect and substitution modelling for reactivity insights
- Spectral predictions (IR/Raman/XAS) for experimental validation
Example Applications
- Ge incorporation in sphalerite
- Apatite surface chemistry for flotation
- CO₂/H₂O adsorption on silicate surfaces for carbonation studies
Outputs Ready for Use
- Tables of reaction and adsorption energies
- Surface constants for geochemical databases
- Speciation files for thermodynamic modelling
- Clear visualisations of structures and adsorption geometries
Tools: VASP · CP2K · VMD
Related: Mineral spectra · AIMD · XFM integration
Previous post:
Coming up next: