Ab initio molecular dynamics
We use ab initio molecular dynamics (AIMD) to uncover reaction mechanisms at mineral–water interfaces and in solution, incorporating explicit H₂O or alternative solvents under varying temperature and pressure conditions. This approach provides dynamic, kinetic insights beyond the reach of conventional static quantum mechanical calculations.
Typical questions
- How do metal–ligand reactions behave in aqueous fluids under varying temperature, pressure, and chemical environments (pH, Eh, salinity)?
- What are the predominant reactions during mineral dissolution and precipitation steps?
- How do ion pairing and solvent structure evolve with changes in temperature, pressure, and salinity?
Approach
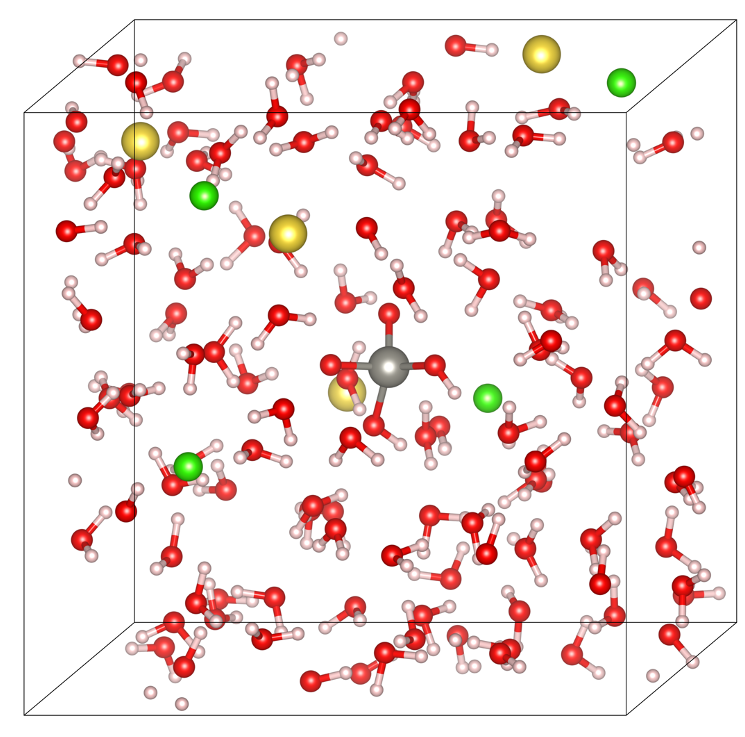
Simulation box of tungsten in hydrothermal fluids
- AIMD Simulations: Perform ab initio molecular dynamics in solvent with trajectories of ~10–100 ps to capture geometry and chemical speciation.
- Enhanced Sampling: Map free-energy surfaces of key chemical reactions for deeper mechanistic insights.
- Thermodynamic Calculations: Predict properties for reactions where experimental data are unavailable.
- Periodic Interface Models: Build realistic mineral–water interface models to study interfacial processes.
Deliverables
- Detailed speciation and molecular geometry across a range of temperature, pressure, and chemical compositions.
- Free-energy surfaces and kinetic descriptors for key reactions.
- Thermodynamic properties for systems and reactions of interest.
- High-quality visualisations for demonstration, reports and publications.
Example applications
- Gold speciation in potassium rich fluids and impact in K-alteration
- Tungsten speciation in hydrothermal fluids
- Cu speciation and thermodynamics in Cl and HS-rich geofluids
- revisit Zn in HS and Cl-rich fluids
- REE complexation and reaction dynamics
Tools
CP2K (GPW), CPMD, VASP (BOMD), post‑processing scripts.
Related
#mineral-spectra · #thermo-rtm
Previous post:
Coming up next: